摘要
miRNA的失调与各种人类疾病的发生和进展密切相关,因此识别疾病相关的miRNA对于研究疾病发生机理至关重要。多数先前的方法是基于同质性假设提出的,即存在相互连接的节点通常具有相似的特征。因此,它们无法充分利用异质网络中多类节点属性和多样连接边的异质性信息。本研究提出了一个新的关联预测方法,HHIE,来编码和学习miRNA-疾病异构网络中的同质性和异质性信息。首先,我们建立基于低频和高频滤波器的同质-异质信息编码策略,来提取相互连接的节点特征的共性,即编码节点间的同质性。再者,该编码策略也针对多类节点的异质性,提取目标节点与多跳邻居节点间的差异性信息。然后,建立残差连接来聚合编码器中多层的输出,以降低传统节点特征聚合过程中容易产生的过平滑问题。最后,设计了局部信息聚合策略来整合目标节点与邻居节点的相似性,进而确定节点的同质性来引导特征融合。五倍交叉验证的实验结果表明,该研究方法HHIE在推断miRNA-疾病关联方面优于几个先进的预测方法。此外,消融实验展示了同质性信息学习、异质性信息编码和原始特征嵌入,对于疾病相关miRNA候选预测性能提升的贡献。
关键词
Abstract
Dysregulation of miRNAs is closely related to the occurrence and progression of all kinds of human diseases. Identifying disease-associated miRNAs plays an important role in investigation of pathogenesis of diseases. Most of the previous approaches were proposed based on the homogeneity assumption, i.e., the nodes with connections are usually more likely to have similar features. Therefore, they did not completely utilize the heterogeneity of the heterogeneous network with multiple types of node attributes and diverse connections. We proposed a novel association prediction method, HHIE, to encode and learn the homogeneity and the heterogeneity in the miRNA-disease heterogeneous network. We presented a strategy for encoding the homogeneity and heterogeneity based on the low-frequency and high-frequency filters. The strategy can extract the common features of nodes which reflect the homogeneity among multiple nodes. Moreover, the encoding strategy may obtain the discriminative features between a target node and its multi-order neighbor nodes. The residual connections are designed to aggregate the features from multiple encoding layers to alleviate the over-smoothing problem of feature aggregation. Finally, we presented a local information aggregation strategy to integrate the similarities between the target node and its neighbors, and then determine the homogeneity of nodes to guide the feature aggregation. The five-fold cross validation experimental results indicated that our method, HHIE, outperformed several state-of-the-art prediction methods for inferring the miRNA-disease associations. In addition, the ablation experiments demonstrated the contribution of homogeneity information learning, the heterogeneity information encoding, and the original feature embedding, to the improved prediction performance.
MicroRNAs(miRNAs)是一类小的非编码RNA,长度约为22~24个核苷酸[1-2]。miRNA的表达异常与多种疾病的发生有关[3-4],因此识别疾病相关的miRNA有助于探索疾病的发病机制,并促进疾病的诊断和治疗。计算预测疾病与miRNA关联的可能性,可以帮助选择每个疾病的候选miRNA,减少生物学实验所需的成本和时间。现有的miRNA-疾病关联预测方法主要包含三大类:
1)第一类方法是基于miRNAs的功能越相似,它们就越有可能与相似疾病关联的生物前提[5]。因此,研究人员通过两个miRNA相关的疾病,来确定miRNA功能相似度,然后根据这些相似度构建miRNA相似网络[6]。Xuan等[7]利用miRNA家族和簇的信息测量miRNA的相似性,并通过前k个最相似的邻居鉴定出潜在的疾病相关miRNA。Chen等和Xuan等[8-9]构建了基于相似性网络随机游走的预测模型。然而这几个方法难于为新的疾病预测它们相关的候选miRNA。
2)第二类方法是引入了额外的疾病相关数据来构建miRNA-疾病异构网,以整合miRNA相似性,疾病相似性和miRNA-疾病关联。Chen等[10]提出了一种基于k个最相似邻居的预测方法以发现miRNA与疾病之间的潜在关联,You等[11]提出了一种深度优先的搜索方法。Wang等[12]将miRNA序列信息引入他们的模型中,并使用逻辑树分类器进行miRNA与疾病的关联预测。某些基于矩阵分解的方法也被用来进行预测疾病相关的miRNA[13-17]。然而,上述方法没有充分利用多种连接之间的差异性。miRNA-疾病关联评分也可以通过随机漫步[18-21]、正则化最小二乘法[22]、miRNA-疾病网络投影[23-24]和标签传播[25]来获得。这些方法构建了浅层预测模型,且大部分方法只关注于聚合相邻邻居的信息,未能挖掘miRNA和疾病节点的异质性。两个相似的miRNA更有可能与相似的疾病关联。根据这个生物学意义,我们可以利用浅层预测模型寻找到疾病的潜在miRNA候选。例如:如果miRNAmir2和miRNAmir3相似,且mir2关联于疾病dis4,那么mir3就可能与dis4关联。这种浅层关联可以通过浅层模型获得。
3)第三类方法是基于深度学习的方法来整合疾病和miRNA相关信息。miRNA-疾病双层异构网中存在多种连接边并且这些连接边存在着非线性的复杂联系,距离较远节点之间的深层关联难以通过“两个相似的miRNA更有可能与相似的疾病关联”的生物学意义这种直观视角得到。通过深度学习的方法可以学习所有miRNA和疾病节点的深层而有代表性的特征,从而进一步评估它们关联的可能性。Peng等[26]和Xuan等[27]设计了基于卷积神经网络的预测模型。Hu等[28]构建了自适应深度传播图神经网络模型来预测疾病相关候选miRNA。一些基于图卷积神经网络(GCN)[29-32]、生成对抗网络[33]、自动编码器[34-37]和深度置信网络[38]的关联预测模型也被建立。这些基于深度学习的模型学习了miRNA和疾病之间的深层联系,并具有更好的预测性能。
总之,多数先前的方法都是基于同质性假设的,它们仅仅聚合来自于相邻节点的信息。我们提出了一个新的预测方法,称为HHIE,通过在miRNA-疾病异构网上设计低通和高通的增强型滤波器引导的编码策略,从而分别收集同质性信息和异质性信息,并利用局部相似性学习权重来进行自适应的融合。
1 材料与方法
本研究提出了一个关联预测模型HHIE来预测与给定疾病相关的候选miRNA,如图1所示。首先构建了miRNA-疾病双层异构网,并通过在异构网上使用低通滤波器和高通滤波器引导的多层编码策略,分别获取了同质性信息和异质性信息。考虑节点的局部拓扑,利用局部相似性学习各个中间表示层信息的权重来聚合来自不同编码层的多种信息,最终得到各节点的多信息融合表示。最后将miRNA-疾病节点对的信息融合表示拼接,并通过全连接神经网络得到miRNA-疾病的关联预测得分。

图1HHIE模型框架
Fig.1Framework of the proposed HHIE model
1.1 相关数据集
人类miRNA-疾病数据库(HMDD)包含7 908个miRNA与疾病的关联[39],它们涵盖了793个miRNA和341个疾病。从美国国家医学图书馆[40]获得疾病的术语信息,并使用它们建立疾病的有向无环图(DAGs)[6]。两个疾病的语义相似性是通过它们的DAGs计算得到的。
1.2 双层异构网构建与矩阵表示
构建了一个miRNA-疾病双层异构网G=(V,E,W),其中节点集V={Vmir∪Vdis},表示Nm个miRNA节点,Vdis表示Nd个疾病节点。权重为wij∈W的边eij∈E连接一对节点vi,vj∈V。对于异构网G,节点之间的连接有两种,一种是相同类型节点之间的层内连接,如miRNA-miRNA相似性连接,疾病-疾病相似性连接。另一种是不同类型节点之间的层间连接,包括miRNA-疾病关联连接。W定义为W=(Asim,Bmir-dis),其中Asim是层内关系矩阵,Bmir-dis是层间关系矩阵。
Asim包括miRNA相似性矩阵和疾病相似性矩阵,定义为
(1)
一个疾病的有向无环图(DAG)通常由该疾病及其相关的疾病术语及它们间的连接边组成[6]。通过Wang等[6]的方法,基于两个疾病disi和disj的DAGs,两者之间的语义相似性Adisij被计算得到。设Ti为的disi术语集,每个疾病术语t在Ti中的语义贡献ξi(t)定义为
(2)
其中disi是最具体的术语,并且其语义贡献被设置为1。同时,其祖先节点越普遍,其语义贡献就越小。疾病disi的总语义值为
(3)
如果第i个疾病disi和第j个疾病disj有更多共同的疾病术语,它们就可能更相似。disi和disj之间的语义相似性定义如下:
(4)
其中ξi(t)(ξj(t))是与disi(disj)相关的第t个术语的语义值。
Amir包含了Nm个miRNA间的相似性。两个miRNA的功能越相似,它们就越有可能与相似的疾病相关。基于这个生物前提并受到Wang等[6]方法的启发,基于miri和mirj关联的两组疾病,计算得到它们的相似性Amirij。例如miRNAmira与一组疾病DTa={disa1,disa2,...,disam}关联,miRNAmirb与一组疾病DTb={disb1,disb2,...,disbn}关联。然后计算DTa和DTb的相似度作为mira和mirb的相似度,
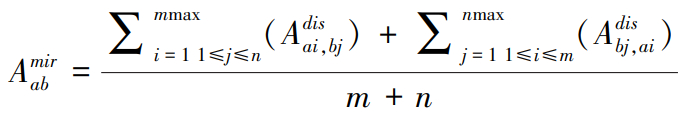
(5)
其中Adisai,bj为疾病disai和disbj的语义相似度,disai∈DTa且disai∈DTb。m和n是DTa和DTb所包含的疾病的数量。Amirij和Adisij的值在0和1之间。Amirij(Adisij)的值越高,表明miri(disi)和mirj(disj)之间的功能(语义)越相似。
Bmir-dis反映了Nm个miRNA和Nd个疾病之间的关联关系,
(6)
其中,Bmir-dis的每一行代表一个miRNA,每一列代表一个疾病。如果miri与disj关联,则Bmir-disij=1,否则Bmir-disij=0。
给定相似性矩阵Amir,Adis和关联矩阵Bmir-dis,得到异构网G中所有边的权重矩阵W,
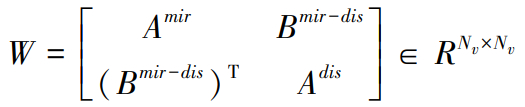
(7)
其中,(Bmir-dis)T是Bmir-dis的转置,并且Nv=Nm+Nd。G中所有节点通过边连接形成邻接矩阵C,C=W。对于miRNA或疾病节点,与之相关的相似性和关联可以视为节点的属性。因此,W被视为中miRNA和疾病节点的属性矩阵,并表示为X。
1.3 同质-异质信息编码
异构网G蕴含了所有miRNA和disease节点之间多样的联系和各种各样的连接,这意味着每个节点都蕴含不同的同质性信息和异质性信息。提出了一个同质-异质信息编码策略,通过低通和高通的增强型滤波器来分别收集异构网中的同质性信息和异质性信息,使模型能同时在异质图和同质图上提供良好的性能。低通滤波器主要保留了多个节点之间特征的共性,使学习到的相互连接节点表示变得更相似。而高通滤波器用于捕获节点之间差异,并且防止仅仅保留节点的共性后会使节点表示将变得无法区分而导致的过平滑问题。同时考虑局部拓扑,利用局部相似性来表示节点级的同质性,最终引导编码器各层中间表示的融合权重的学习。
1)节点vi的同质性。节点vi的同质性H(vi)是与节点vi具有相同类型的邻居数与节点vi的邻居数之比,表示为
(8)
其中是节点vi的邻居节点集。
2)图G的同质性。图G的同质性H(G)是所有节点同质性的平均值,表示为
(9)
3)节点vi的异质性用1-H(vi)来度量,图的异质性为1-H(G)。它们的值都在0~1之间。
设计了有L个编码层的同质-异质信息编码策略来分别捕捉节点的多样的信息。在每一个编码层,利用添加加权单位矩阵I(即加权自环)的低通和高通的增强型滤波器来分别学习异构网中节点的同质性信息和异质性信息。低通滤波器过滤低频信号[41],聚合低频信息,忽略节点之间的异质性。高通滤波器过滤高频信号,表示节点特征与邻域节点特征的差异,如图2所示。两个滤波器分别表示为
(10)
(11)
其中Fhom是低通滤波器,Fhet是高通滤波器且Fhom+Fhet=I。邻接矩阵,D表示度矩阵且。

图2低频、高频信息的过滤
Fig.2Filtering of low-frequency and high-frequency information
同时,设计使用初始残差连接来聚合不同层的输出,以获得有更多信息的中间表示,防止简单聚合上一层的表示而产生的过平滑问题。为了利用来自初始节点特征的所有信息,将原始输入中从未处理过的信息提供给下一层。第一层和第层同质-异质信息编码层的输出可以表示为
(12)
(13)
其中,FH是滤波器,H∈{hom,het},μ是超参数且μ∈[0,1],可以看作是前?-1层提取的总信息,=2,3,...,L。
经过L个同质-异质信息编码层,分别得到同质信息表示矩阵,和异质信息表示矩阵。将它们分别投影到同一低维空间,表示为
(14)
(15)
其中,θ是激活函数Relu,是权重矩阵。为了利用初始节点特征,也对X进行投影得到,
(16)
其中,θ是激活函数Relu,WI是权重矩阵。
1.4 局部信息聚合策略
与目标节点同类型的邻居节点越多,目标节点的同质性越高,反之节点的异质性越高。将局部相似性作为节点的同质性指标[42]。节点的vi的局部相似度Si为
(17)
(18)
其中,是节点vi的邻居节点集,MLPloc:R2→R1是两层感知器。最终所有节点的局部相似性矩阵为S,。
如图3所示,利用局部相似性引导,以及融合,=1,2,3,...,L。它们对最终节点表示的学习贡献不同,所以将S作为局部拓扑信息,并同时使用S,S2来引入非线性并得到不同通道的节点表示的权重:
(19)
其中是一个两层感知机。所以节点的第L层的多通道信息融合表示计算如下:
(20)
其中,⊙表示元素相乘。最后将原始矩阵和每一层的多通道信息融合表示矩阵拼接得到所有节点的同质-异质信息融合表示矩阵Z,表示为
(21)
其中‖表示连接操作。

图3局部信息聚合过程
Fig.3Process of local information aggregation
1.5 最终整合和预测
给定同质-异质信息融合表示矩阵Z,其中miRNA节点miri的同质-异质信息融合表示为Z(i),疾病节点disj同质-异质信息融合表示为Z(Nm+j)。将Z(i)和Z(Nm+j)左右连接形成K。K经过一个全连接层和softmax层,可以获得miri和disj的关联预测分数score,
(22)
其中Wmul和bmul是可学习的权重矩阵和偏差向量。
将交叉熵作为关联预测分数score与实际标签t之间的损失函数并使用Adam算法进行优化,定义为
(23)
其中Ntra是训练样本集的数量。当miri和disj之间存在实际关联时,tj=1,否则tj=0。
2 实验结果与分析
2.1 参数设置和评价指标
在同质-异质信息编码模块中,编码层数被设置为3,超参数=0.5,并且μ=0.5。同质-异质信息编码层的输出维度为1 134,经过投影以后矩阵的输出维度为400。HHIE基于深度学习框架PyTorch实现,训练轮数设置为100,学习率为0.000 5。
五倍交叉验证被用来评估HHIE和其他用于推断miRNA相关疾病模型的性能。所有已知的miRNA-疾病关联被作为正样本,其中4/5用作训练,剩余的用于测试。负样本由所有未观察到的miRNA-疾病关联组成。随机选择与训练的正样本数目相等的负样本,添加到训练集中,剩余的负样本用于测试。以受试者操作特征(ROC)曲线下面积(AUC)[43],精确召回率(PR)曲线下面积(AUPR)[44]作为评估指标。一方面,在miRNA-疾病关联预测问题中,反例样本和正例样本的比例约为33∶1,于是它们之间存在严重的类别不平衡。在类别不平衡的情况下,AUC通常比准确率等指标更能提供准确的评估;另一方面,在类别不平衡时,Precision-recall曲线下的面积(AUPR)通常比AUC能提供更具信息含量的评估结果。因此,使用AUC和AUPR来评估miRNA和疾病关联的预测方法。在五倍交叉验证过程中,首先计算每倍的平均AUC和AUPR,最终得到五个结果的平均值。
2.2 消融实验
利用消融实验来验证异构网中学习到的同质性信息、异质性信息以及原始特征信息对预测miRNA-disease关联的贡献。如表1所示,最终模型HHIE取得了最好的性能。如果不使用低通滤波器学习同质性信息,与最终模型相比,AUC下降了0.5%,AUPR减少了0.6%。它在AUC方面取得了第二好的性能,从生物学视角来看,可能的原因是:对于两个相似性更高的miRNA,它们更有可能与类似的疾病相关。类似的,两个更相似的疾病也会与同样的miRNA有较高的关联。于是设计低通滤波器学习近距离节点之间更相似特征是必要的。在不使用高通滤波器学习异构网中异质性信息的情况下,AUC和AUPR分别比最终模型低0.4%和2.3%。结果表明,高通滤波器对AUPR的影响是第二大的。这表明高通滤波器可以有效地捕捉较远距离节点之间的差异性从而对AUPR的提升有较大的贡献。与不学习原始节点特征的模型相比,最终模型在AUC和AUPR中的性能分别提高了0.8%和3.2%。原始特征对预测性能提升的贡献最大,因为它包含了各个节点和所有其它miRNA-疾病节点之间的直接相似性和关联信息和更多的细节信息。
表1消融实验的结果
Table1Results of ablation experiments

2.3 与其他方法的比较
HHIE与八种最先进的miRNA-疾病关联预测方法进行比较,包括GMDA[33],AEMDA[34],DBNMDA[38],NCMCMDA[16],GSTRW[21],DMPred[19],PBMDA[11]和 Liu等[19]的方法。使用HHIE和这些方法各自的最佳参数设置来报告它们的最佳性能。下面对几个比较方法进行了简要描述:
1)GMDA:它整合了miRNA和疾病节点对的邻居拓扑,miRNA家族和簇的属性信息,并基于生成对抗网络预测miRNA-疾病关联。
2)AEMDA:AEMDA建立了一个基于全连接的自动编码器的模型来推断miRNA和疾病关联的可能性。
3)DBNMDA:它深度整合了miRNA与疾病之间的复杂关系,建立了基于深度置信网络的模型。
4)NCMCMDA:它是基于矩阵完成的方法,将邻域约束与矩阵填充相结合并充分利用了相似性信息。
5)Liu的方法:该方法整合多个数据源建立了疾病相似性网络和miRNA相似性网络,并构建了miRNA-疾病异构网。该预测模型是在异构网上进行随机游走的得到的。
6)DMPred:它面向miRNA-疾病双层异构网络建立了基于非负矩阵分解的预测模型。
7)PBMDA:该模型整合了miRNA-miRNA相似性、疾病-疾病相似性和miRNA-疾病关联数据,建立了基于深度优先搜索算法的预测模型。
8)GSTRW:它整合了所有miRNA(疾病)之间的全局相似性以及通过在miRNA网络和疾病网络上随机行走获得的信息。
HHIE和被比较的方法使用相同的训练数据集和测试数据集进行训练和测试。9个方法的关于341个疾病的ROC曲线和PR曲线如图4所示。对所有疾病的平均AUC而言,HHIE得出了最高AUC,为0.932,它的表现优于GMDA 0.4%,比Liu的方法好4.1%,优于PBMDA 7.5%,比DMPred和DBNMDA高4.2%和2.5%,超过NCMCMDA 2.7%, AEMDA 1.6%,优于性能最差的GSTRW 12.5%。同样的,HHIE获得了最高的AUPR值(AUPR=0.275),比GMDA,PBMDA,DBNMDA,Liu的方法、GSTRW,NCMCMDA,AEMDA和DMPred分别高出2.5%、18.5%、8.8%、17.6%、23.5%、10.9%、5.6%和18.9%。综上所述,HHIE取得了最佳性能,GMDA和AEMDA的表现位于第二、第三位。对于GMDA,它基于生成对抗网络,集成了miRNA和疾病节点对的邻居拓扑。AEMDA使用一个深度自动编码模型来学习潜在关联。DBNMDA的方法基于深度信念网络,深度整合了miRNA与疾病之间的复杂关系。它们都忽略了异构网中节点的异质性信息,即目标节点和邻居节点的差异性。NCMCMDA基于矩阵完成的方法,并充分利用了相似性信息。Liu的方法和DMPred的AUC和AUPR值相近,两者分别建立了基于随机游走和非负矩阵分解的模型。PBMDA的AUPR略高于DMPred,但其AUC比DMPred低3.3%。GSTRW的表现比HHIE差的多。GSTRW整合了所有miRNA(疾病)之间的全局相似性以及通过在miRNA网络和疾病网络上随机行走获得的信息。然而,这是一个浅层的预测模型,该模型很难学习miRNA和疾病节点之间深层复杂的特征。大多数比较方法是基于图卷积网络、图神经网络和卷积神经网络提出的。它们都属于深度学习方法,可以挖掘miRNA和疾病节点的深层特征。实验结果表明,这些方法比GSTRW具有更好的预测性能,这表明学习节点的深层特征是必要的。HHIE相较于其他方法的改进主要是因为同时考虑到目标节点与邻居节点的相似性和差异性,学习到节点的同质性信息和异质性信息。

图4不同预测方法的ROC曲线与PR曲线
Fig.4ROC curves and PR curves of different prediction methods
注:(a)ROC curves;(b)PR curves.
2.4 案例分析
为了进一步证明HHIE发现潜在miRNA-disease相关性的能力,对3个疾病进行了案例研究,包括乳腺肿瘤、肺肿瘤和卵巢肿瘤。以乳腺肿瘤为例,通过模型获得其候选miRNA的关联得分,并按降序排列,收集前50个候选miRNAs进行分析。
名为miRCancer的数据库包含经实验验证的9 080个miRNAs与疾病之间的关联,这些关联是Xie等[45]利用文本挖掘技术从已发表的文献中提取的,并经过了进一步手动确认。miR2Disease也是一个数据库,包含了349个miRNAs,163个疾病,3 273个miRNAs-疾病相关信息。如表2所示,乳腺肿瘤的20个候选miRNAs被miRCancer收录,5个候选miRNAs被miR2Disease收录。这表明这些miRNAs在乳腺肿瘤中的表达上调或下调。
dbDEMC是一个集成数据库,存储了通过高通量和低高通量方法检测到的疾病中的差异表达miRNA[46]。该数据库收录了2 224个miRNAs,36个肿瘤。dbDEMC包括44个候选miRNAs,它们在乳腺细胞中表达异常。在50名候选中,有1个候选miRNA未被观察到的证据证实,被标记为"unconfirmed"。
表2乳腺肿瘤相关的前50个候选miRNAs
Table2The top 50 miRNA candidates related to breast neoplasms

表3记录了卵巢肿瘤的50个候选miRNAs,miRCancer和dbDEMC分别包含26个和49个候选miRNAs,表明了这些miRNAs可能参与了卵巢肿瘤的发展。在肺肿瘤的50个候选miRNAs中(表4),miRCancer记录了40个候选miRNAs,dbDEMC和miR2Disease分别记录了48和16个候选miRNAs,说明这些miRNAs确实与肺肿瘤有关。所有的案例研究表明,HHIE确实能够发现潜在的候选miRNA-疾病关联。
表3卵巢肿瘤相关的前50个候选miRNAs
Table3The top 50 miRNA candidates related to ovarian neoplasms

表4肺肿瘤相关的前50个候选miRNAs
Table4The top 50 miRNA candidates related to lung neoplasms

3 结论
1)我们提出的新的方法HHIE可以有效地进行miRNA-疾病关联预测。5倍交叉验证结果表明,HHIE的AUC和AUPR均高于对比的方法,取得了更好的预测性能。通过对包括乳腺肿瘤、肺肿瘤和卵巢肿瘤在内的3个疾病的案例研究,HHIE在发现潜在的miRNA-疾病关联方面的能力得到了展示;
2)我们的方法HHIE和一些对比的方法还有一定的局限性。一个没有已知相关疾病信息的miRNA,不能加入到异构网中。因此,那些利用基于相关疾病计算得到的miRNA相似性数据的预测方法,无法预测这些疾病相关的候选。具有局限性的方法包括我们的方法,也包括我们对比的GMDA、Liu的方法、PBMDA、AEMDA。根据miRNA相关的靶基因来计算miRNA间的相似性,可以作为那些没有关联疾病的miRNA节点的补充信息。因此,在未来的工作中,我们打算深度整合基于靶基因的miRNA相似性和基于疾病的miRNA相似性,从而建立更全面且有效的预测模型。