 2023, Vol. 21
2023, Vol. 21


序列比对是指利用计算机算法和程序,比较两个或多个核酸或蛋白质一级结构核苷酸或氨基酸的异同。序列比对的研究对象为核酸序列或蛋白质序列。构成核酸序列的基本单元为核苷酸,而构成蛋白质序列的基本单元为氨基酸。为深入分析序列比对结果,理解序列比对的生物学意义,对构成核酸和蛋白质序列的基本单元需要有一个基本的了解。
1.1 核酸序列基本单元通常所说的核酸包括脱氧核糖核酸(Deoxyri-bonucleic Acid, DNA)和核糖核酸(Ribonucleic Acid, RNA)两大类。DNA序列的基本单元为脱氧核糖核苷酸(Deoxyribonucleotide),由脱氧核糖核苷(Deoxyribonucleoside)和磷酸(Phosphoric Acid)组成。脱氧核糖核苷包括脱氧核糖和碱基两部分。碱基分两类,一类为嘌呤碱(Purine),简称嘌呤,包括腺嘌呤(Adenine, A)和鸟嘌呤(Guanine, G)两种; 另一类为嘧啶碱(Pyrimidine),简称嘧啶,包括胞嘧啶(Cytosine, C)和胸腺嘧啶(Thymine, T)两种(见图 1)。

|
图 1 DNA分子中四种脱氧核糖核苷酸分子结构及相互之间的关系 Figure 1 Four types of deoxyribonucleotide in DNA and their relationships |
显然,四种不同碱基决定了四种不同脱氧核糖核苷酸。为便于表述,脱氧核糖核苷酸经常简称核苷酸,分别用四种不同核苷酸的英文首字母,即腺嘌呤Adenine用A表示,鸟嘌呤Guanine用G表示,胸腺嘧啶Thymine用T表示,胞嘧啶Cytosine用C表示。习惯上所说“DNA分子由四种不同碱基组成”,实际上是指由含不同碱基的四种核苷酸组成,更确切地说,应该是由含不同碱基的四种脱氧核糖核苷酸组成[1]。
DNA分子由四种不同核苷酸通过磷酸键按一定顺序首尾相接,即前1个核苷酸糖环第3’位碳原子与下一个核苷酸糖环第5’位碳原子通过磷酸二酯键相连,这就是通常所说的DNA序列一级结构。
显然,磷酸二酯键的连接方式,决定了DNA序列具有方向性。DNA分子在体内合成时,由四种不同碱基A, G, T, C按5’到3’方向不断延伸、依次排列,其产物为不同长度、不同顺序的DNA分子。显然,四种碱基的排列顺序不同,所形成的DNA序列的一级结构也就不同。地球上除RNA病毒外的其它生物,包括动物、植物、细菌和DNA病毒,其遗传信息均取决于其DNA序列。
1953年,沃森和克里克提出DNA分子双螺旋模型,即DNA分子由两条方向相反的互补链构成,四种不同碱基通过氢键配对原则,即A和T配对、G和C配对,形成螺旋形的二级结构。DNA分子双螺旋模型,从分子水平上揭示了遗传信息复制和传递的机制。
分析四种不同碱基的结构可以发现,它们之间具有一定关系。若用正四面体的四个顶角分别表示A, G, C, T四种核苷酸,正四面体的六条边则可以表示四种核苷酸之间的关系(见图 1b),分别用字母R, Y, M, K, W和S表示。根据国际理论和应用化学联合会(The International Union of Pure and Applied Chemistry,IUPAC)、国际生物化学和分子生物学联合会(The International Union of Biochemistry and Molecular Biology,IUBMB)制定的核苷酸代码(见表 1),R表示嘌呤A或G,Y表示嘧啶C或T; M表示含氨基的腺嘌呤A或胞嘧啶C,K表示含酮基的鸟嘌呤G或胸腺嘧啶T; W表示能够形成两对氢键的腺嘌呤A或胸腺嘧啶T,意为弱耦合(Weak),S表示能够形成三对氢键的鸟嘌呤G或胞嘧啶C,意为强耦合(Strong)。
| 表 1 核苷酸名称和代码 Table 1 Name and code of nucleotide |
此外,N表示四种核苷酸中的任意一种,在基因组序列中,通常用来表示无法测定或测定结果不能确定的位点。
了解核苷酸代码,不仅在序列比对中,而且在限制性内切酶分析和简并引物设计等序列分析实际应用中,都很有帮助。
1.2 蛋白质序列基本单元蛋白质序列基本单元为氨基酸。常见氨基酸有二十种,氨基酸的基本结构包括主链和侧链两部分。不同氨基酸的主链相同,而侧链不同。按侧链基团大小、亲疏水性和电荷性等不同性质,可以将它们分成四大类(见图 2)。

|
图 2 二十种常见氨基酸名称和分类 Figure 2 Name and classification of 20 common amino acids |
第一类为疏水氨基酸,包括丙氨酸(Ala, A)、缬氨酸(Val, V)、亮氨酸(Leu, L)、异亮氨酸(Ile, I)、甲硫氨酸(Met, M)五种侧链为脂肪族基团的氨基酸,以及侧链为芳香族的苯丙氨酸(Phe, F)。第二类为带电氨基酸,根据电荷性质的不同,又可分为带负电的门冬氨酸(Asp, D)和谷氨酸(Glu, E),带正电的组氨酸(His, H)、赖氨酸(Lys, K)和精氨酸(Arg, R)。第三大类则是既不疏水、又不带电的极性氨基酸,其中丝氨酸(Ser, S)、苏氨酸(Thr, T)和酪氨酸(Tyr, Y)侧链具有极性羟基,而门冬酰胺(Asn, N)和谷氨酰胺(Gln, Q)的侧链具有酰胺基,另外一个色氨酸(Trp, W)比较特殊,其侧链为吲哚环,也属于极性氨基酸。最后一类包括半胱氨酸(Cys, C)、脯氨酸(Pro, P)和甘氨酸(Gly, G)三个氨基酸。其实,这三个氨基酸性质各不相同,很难将它们归到其它类别中,将它们归为同一类,只是为了便于记忆。
除了亲疏水性、极性和电荷性三种性质外,二十种氨基酸的溶剂可及性(Solvent Accessibility)、刚性(Bulkiness)、跨膜倾向性(Transmembrane Tendency)和可突变性(Mutability)也各不相同(见表 2)。
| 表 2 二十种氨基酸基本性质 Table 2 General properties of 20 amino acids |
表中不同氨基酸的性质源自以下几个网站,有兴趣的读者可自行查看。
1) 氨基酸结构特性(Amino Acid Property)
德国海德堡大学蛋白质结构、功能和演化研究组氨基酸结构特性网站:
http://www.russelllab.org/aas/aas.html
2) 蛋白质序列波形图(ProtScale)
瑞士生物信息研究所蛋白质分析专家系统(Expert of Protein Analysis System, Expasy)蛋白质序列特征波形图网站:
https://web.expasy.org/protscale
3) 蛋白质序列特征参数(ProtParam)
Expasy蛋白质序列特征参数网站:
https://web.expasy.org/protparam
氨基酸种类的多样性,是自然界长期演化的结果,它决定了蛋白质功能的多样性,从而也决定了生物多样性。必须说明,将不同氨基酸分为亲水/疏水、极性/非极性、带电/不带电等, 仅仅是为了便于理解,并没有绝对的界限[2]。例如,赖氨酸侧链末端带正电荷,但其侧链包含四个疏水性甲基。又如,组氨酸侧链带正电,通常分布在分子表面; 但在某些转录因子中,组氨酸常与半胱氨酸一起,与锌原子组成锌指结构,分布在分子内部,具有疏水特性。
2 序列比对基础 2.1 相似性和同源性序列比对是指利用计算机程序比较核酸或蛋白质序列之间相似性,找出两个或多个序列之间的相同区域或差异位点。根据分子生物学中心法则,DNA是遗传信息携带者,而蛋白质则是功能分子。不同物种之所以千姿百态、各不相同,其内在原因是它们的基因组不同,或者更确切地说,是它们的DNA序列及其编码所得的蛋白质不同。
序列比对经常用来判断所比对的两个序列是否为同源序列。必须指出,相似性(Similarity)和同源性(Homology)是两个完全不同的概念。根据达尔文进化论学说,地球上现有物种,不论是动物、植物,或者是微生物,可以追溯到一个共同祖先。由于地球环境不断变化,祖先物种在演化过程中发生分化,形成新物种,以适应变化后的新环境。演化过程中,不同物种的基因组核酸序列及其编码的蛋白质序列均发生不同程度的突变,但依然保持一定的相似性。
序列相似性概念在核酸和蛋白质序列中有所不同。核酸序列的相似性高低,是指通过序列比对所得结果中相同核苷酸残基所占比例,通常用百分比表示。而蛋白质序列比对结果中,除了用相同氨基酸残基所占比例作为相似性指标外,也经常用相同氨基酸加上相似氨基酸作为相似性指标。所谓相似氨基酸,是指侧链基团理化性质相似的一对氨基酸,如疏水氨基酸亮氨酸(Leu, L)和异亮氨酸(Ile, I),极性氨基酸丝氨酸(Ser, S)和苏氨酸(Thu, T)、带负电的氨基酸门冬氨酸(Asp, D和谷氨酸(Glu, E)、带苯环的氨基酸苯丙氨酸(Phe, F)和酪氨酸(Tyr, Y)等。
不论是核酸序列还是蛋白质序列,序列相似性是指相同和相似残基所占全长序列的比例,比例越高,相似性越高。而序列同源性是指所比较的两个序列是否具有共同的祖先序列。显然,序列同源性只有是非之别,没有高低之分,所谓“具有50%同源性”,或“高度同源”等说法,都是错误的。值得一提的是,相似性和同源性的概念之所以容易混淆,是因为两者之间关系密切。一般说来,同源序列特别是亲缘关系较近的序列,相似性通常较高; 反之,相似性较高的两条序列,很有可能具有共同祖先。也就是说,序列相似性的高低经常用来推断其是否同源。
同源序列通常分为直系同源(Ortholog)和并系同源(Paralog)两类。以血红蛋白为例,小鼠和大鼠两个不同物种alpha血红蛋白在它们共同祖先中已经存在。随着物种分化,小鼠和大鼠共同祖先分化为两个物种,所形成的新物种通过遗传机制获得祖先物种基因组中alpha珠蛋白基因。因此,小鼠和大鼠两个不同物种中alpha血红蛋白称为直系同源蛋白,其编码基因则为直系同源基因。而小鼠中alpha珠蛋白基因在物种形成后,由基因复制产生了两个基因,编码两个alpha血红蛋白,即alpha1和alpha2。小鼠血红蛋白alpha1和alpha2则称为并系同源(有时也译作旁系同源)蛋白,其编码基因则称为并系同源基因。
2.2 整体比对和局部比对双序列比对的方法可以分为两种,一种从全长序列出发,考虑所比对的两条序列的整体相似性,即整体比对(Global Alignment); 另一种仅考虑所比对序列部分区域的相似性,即局部比对(Local Alignment)。一般说来,亲缘关系近的物种间的序列相似性较高,而且经常具有整体相似性; 而亲缘关系较远的物种间序列相似性较低,有时仅有局部相似性。整体比对常用来考察两条序列是否在整体上具有较大相似性,并由此推测它们是否具有同源性。而局部比对则可以找出两个序列中的保守序列片段,如蛋白质序列中某个结构域或功能位点,基因上游启动子区域核酸序列调控元件等。
20世纪70年代初,Needleman和Wunsch提出整体比对算法,即Needleman-Wunsch算法[3]; 80年代初,Smith和Waterman在此基础上提出了局部比对算法,即Smith-Waterman算法[4]。这两种算法是序列相似性比对的基础,至今仍广为使用。
2.3 动态规划和启发式算法以上介绍双序列比对的两种方法,无论是整体比对还是局部比对,都是采用动态规划算法。动态规划算法是指在给定计分矩阵和空位罚分的条件下,通过插入适当空位,使比对结果的总分值最高,即找到最优解。无论是Needleman-Wunsch算法或者是Smith-Waterman算法,都采用计算机领域中常用的动态规划(Dynamic Programming)算法。动态规划算法的核心思想,是把一个复杂问题分解为若干子问题,并通过寻找子问题的解,最终找到初始复杂问题的解。
下面,我们介绍另一种双序列比对方法,即启发式(Heuristic)算法。序列相似性数据库搜索软件Basic Local Alignment Search Tool(BLAST)则采用启发式算法。BLAST通常用于搜索某个蛋白质或核酸序列数据库中与检测序列具有一定相似性的靶标序列。
BLAST算法大体分为以下三步。首先,将检测序列按一定字长(Word Size)拆分成种子(Seed)序列,并按给定计分矩阵和设定阈值,找到与种子序列相似性较高的近邻(Neighbor)序列。接着,逐个找到各近邻序列在数据库中匹配序列,并按分值增加原则向两边延伸,得到高分对(High Scoring Pair)。将所得主对角线方向距离较近的高分对连接起来,并用Smith-Waterman方法进行比对。最后,对搜索到的靶标序列进行统计检验,输出期望值(Expect Value)低于设定阈值的靶标序列,即搜索结果。BLAST也可用于双序列比对,只要把所要搜索的数据库设定为另一个序列。显然,由于所采用的比对策略完全不同,基于Smith-Waterman动态规划算法的比对结果和基于BLAST启发式算法的比对结果不一定相同,某些情况下差别很大。
2.4 计分矩阵和空位罚分无论整体比对还是局部比对,无论采用动态规划算法还是采用启发式算法,都离不开计分矩阵和空位罚分。所谓计分矩阵,是指比对过程中相同或不同核苷酸或氨基酸之间的匹配或错配分值。例如,核酸序列比对时通常匹配分值为正值,而错配分值为负值。蛋白质序列比对时,匹配分值为正值,而错配分值则与氨基酸性质有关,性质不同的氨基酸之间的错配分值为负值,而性质相似的氨基酸之间的分值有可能为正值。不同计分矩阵具有不同匹配分值和错配分值。本文下一节详细介绍核酸序列比对中常用的DNAfull计分矩阵,以及蛋白质序列比对中常用的BLOSUM62和PAM250计分矩阵。显然,计分矩阵不同,比对结果很可能不同。这一点,实际使用时经常被忽略。
序列比对的过程就是利用一定算法或策略,确定是否插入空位、何处插入空位、插入几个空位。这一看似简单的过程,在计算机领域实际上是个难度极大的复杂计算问题,而其背后的生物学机制则更加复杂。所谓空位罚分,是指比对过程中在适当位置插入空位,使比对总分值更高、比对结果更好。实际比对时,程序通常给定默认值,而用户可以根据具体情况进行调整。空位罚分大小设置通常采用经验值,起始空位罚分较大,而延伸空位罚分较小。所谓起始空位,是指插入的第一个空位,而延伸空位则是当插入多个空位时,第二个空位开始的其它空位。此外,当两个长度差别较大的序列进行整体比对时,往往需要考虑是否对末端空位也进行罚分。
2.5 常用软件和分析平台基于动态规划的双序列整体比对算法上世纪七十年代初就已经提出,八十年代初又发表了基于动态规划的局部比对算法。之后不久,随着计算机技术的快速发展,计算机在分子生物学中开始得到应用,双序列比对算法很快用计算机程序实现。上世纪九十年代,在欧洲分子生物学网络组织(European Molecular Biology Network, EMBnet)的支持下,英国学者Peter Rice和Alan Bleasby领导的团队开发了欧洲分子生物学开放软件包(European Molecular Biology Open Software Suite, EMBOSS)。该软件包中包括多个双序列比对程序,其中最为常用的是整体比对程序needle,局部比对程序water,以及基于点阵图的dottup和dotmatcher等。EMBOSS软件包基于Linux系统开发,可免费下载安装在Linux服务器上,用命令行方式运行程序[8]。
为方便用户,欧洲生物信息学研究所(European Bioinformatics Institute, EBI)把EMBOSS软件包中部分常用程序部署在服务器上,包括双序列比对程序needle和water等。而荷兰瓦赫宁根大学的EMBOSS Explorer将整个EMBOSS软件包部署在服务器上,不仅用于双序列比对,还可进行序列格式转换、酶切图谱分析、密码子分析、蛋白质二级结构预测和跨膜螺旋分析等。而美国国家生物技术信息中心(National Center for Biotechnology Information, NCBI)的BLAST分析平台也提供了基于Needleman-Wunsch算法的整体比对工具Global Align和基于启发式算法的Blast2Seq工具。此外,瑞士生物信息研究所(Swiss Institute of Bioinformatics, SIB) 开发的点阵图可视化分析平台Dotlet操作方便、结果直观,可用于重复区域识别、核酸序列中互补序列显示和外显子查找等。为方便用户,下面列出国际知名生物信息中心常用双序列比对工具网址。
·BLAST分析平台
https://blast.ncbi.nlm.nih.gov/Blast.cgi
包括基于Needleman-Wunsch算法的整体比对程序Global Align和基于启发式算法的程序BLAST2Seq,比对结果按BLAST分析平台输出格式展示,包括简要说明(Description)、图形梗概(Graphics Summary)、比对细节(Alignments)和点阵图(Dot Plot)等。
·EBI EMBOSS
https://www.ebi.ac.uk/Tools/psa/emboss_needle
https://www.ebi.ac.uk/Tools/psa/emboss_water/
包括整体比对程序needle和局部比对程序water,两者具有相同的用户界面,包括序列输入和参数设置,可根据需要选择BLOSUM系列或PAM系列不同计分矩阵,并可设置起始和延伸空位罚分。
·荷兰瓦赫宁根大学生物信息中心EMBOSS Explorer
https://www.bioinformatics.nl/emboss-explorer
可进行needle整体比对和water局部比对。
·北京大学生物信息中心网上实验室WebLab
·瑞士生物信息研究所点阵图网站Dotlet
https://myhits.sib.swiss/util/dotlet/doc/dotlet_examples.html
3 常用计分矩阵 3.1 DNAfull核酸序列比对所用计分矩阵与软件或分析平台有关。DNAfull是常用计分矩阵之一。该矩阵可从生物信息学自学网站(http://rosalind.info/glossary/dnafull)下载。本文做了适当改编(见表 3)。
| 表 3 核苷酸计分矩阵DNAfull Table 3 Nucleotide scoring matrix DNAfull |
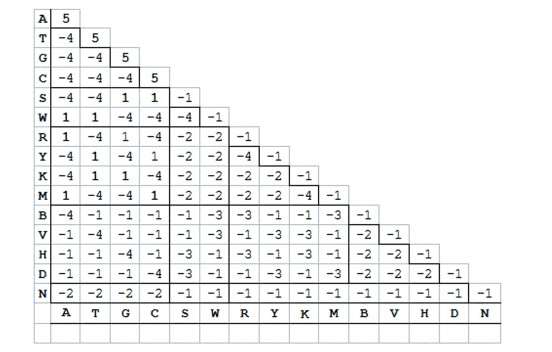
首先,由于该矩阵元素沿主对角线对称分布,原始矩阵中主对角线右上方的元素不再列出。其次,根据四种核苷酸的类别将它们分组。第一组为A、T、G、C四种确定的核苷酸,匹配分值为5,错配分值为-4。若比对序列中仅有这四种核苷酸,不包含歧义核苷酸,则采用上述分值,也可选择另一种计分矩阵DNAmatrix。
实际比对过程中,由于测序精度等原因,有的序列中包含尚不确定的位点,通常用N表示; 而有的序列中包括S、W、R、Y、K、M等,分别表示不同类别核苷酸(见表 1)。例如,S表示G或C两种能形成三对氢键的核苷酸,与G或C的错配分值均为1,而与A或T的错配分值为-4。同理,W表示A或T两种只能形成两对氢键的核苷酸,与A或T的错配分值均为1,而与G或C的错配分值为-4。需要注意的是,S自身匹配分值为-1,低于S与G或C的错配分值。同样,N与四个确定核苷酸的错配分值为-2,而与S、W等不确定核苷酸的错配分值为-1,从某个侧面反映了四种核苷酸的性质及其相互之间的关系。
3.2 BLOSUM62用于蛋白质序列比对的计分矩阵有多种,目前最常用的是BLOSUM62矩阵,也是许多序列比对程序的默认计分矩阵[5]。BLOSUM英文全称为Blocks Substitution Matrix,通常翻译为“模块替换计分矩阵”,后面的数字62表明该矩阵是BLOSUM系列矩阵中的一个。BLOSUM62矩阵可从生物信息学自学网站(http://rosalind.info/glossary/blosum62)下载,本文做了适当改编(见表 4)。
| 表 4 相似性计分矩阵BLOSUM62 Table 4 BLOSUM62 scoring matrix |
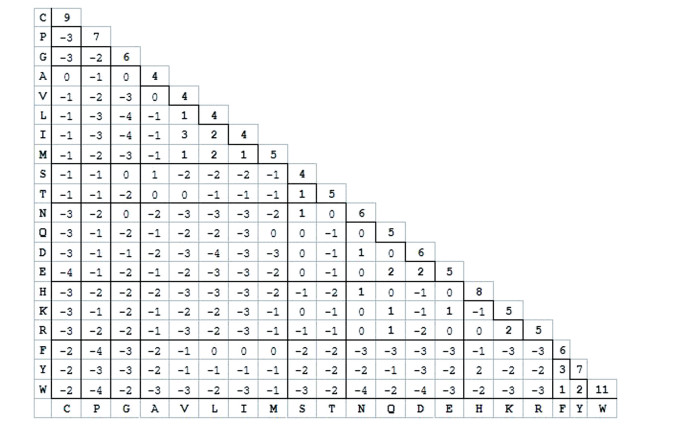
与核苷酸计分矩阵DNAfull类似,主对角线右上方的元素不再列出。基于侧链性质将二十种氨基酸分组,分组原则与图 2氨基酸分类基本一致。五个疏水脂肪族氨基酸丙氨酸A、缬氨酸V、亮氨酸L、异亮氨酸I和甲硫氨酸M分在一组; 两个侧链带羟基的氨基酸(丝氨酸S和苏氨酸T)分在一组; 门冬酰胺N和谷氨酰胺Q、门冬氨酸D和谷氨酸E四个氨基酸分在一组; 带正电的三个氨基酸组氨酸H、赖氨酸K和精氨酸R分在一组; 三个芳香族氨基酸(苯丙氨酸F、酪氨酸Y和色氨酸W)分在一组; 半胱氨酸C、脯氨酸P和甘氨酸G性质独特,各自单独分在一组。需要说明的是,酪氨酸侧链也有羟基,这一点与丝氨酸S和苏氨酸T接近,但其侧链苯环与苯丙氨酸F更加相似,因此将它们分在一组,同组的还有另一个芳香族氨基酸W。
BLOSUM62计分矩阵主对角线的20个矩阵单元为相同氨基酸之间的分值,即匹配分值。不同氨基酸的匹配分值有高有低,如色氨酸W为11、半胱氨酸C为9; 有的较低,如四个脂肪族氨基酸(丙氨酸A、缬氨酸V、亮氨酸L、异亮氨酸I)和丝氨酸S均为4。匹配分值的高低与该氨基酸的性质与丰度有关,也从某个侧面反映了该氨基酸的保守性(见表 2)。分值越高,保守性越强,越不容易发生替换。
除主对角线外的其它矩阵单元为不同氨基酸之间的替换分值,即错配分值。错配分值有正有负,范围在3到-4之间,其中大部分为零或负值。错配分值的高低与两个氨基酸之间的性质有关。两者之间性质差别越大,越不容易发生替换,错配分值也就越低,如第一列半胱氨酸C与谷氨酸E、最后一行色氨酸W与脯氨酸P之间的错配分值均为-4。同组内氨基酸的错配分值相对较高,有的为正值,如缬氨酸V和异亮氨酸I错配分值为3,亮氨酸L和异亮氨酸I的错配分值为2,这是因为它们侧链比较相似,容易发生替换。
以上我们简单介绍了BLOSUM62计分矩阵的特点,以便读者在实际使用过程中深入分析序列比对结果。BLOSUM62是BLOSUM系列计分矩阵中的一个。BLOSUM系列计分矩阵于上世纪九十年代基于蛋白质序列模块数据库BLOCKS构建。除BLOSUM62外,另有BLOSUM30、BLOSUM35、BLOSUM40一直到BLOSUM100共15个,可从NCBI下载(ftp://ftp.ncbi.nlm.nih.gov/blast/matrices)。除BLOSUM62外,其它矩阵的末尾数字均为5或10的倍数。一般说来,BLOSUM100、BLOSUM90等用于相似性高的近缘物种之间的序列比对,而BLOSUM30、BLOSUM35等则用于相似性较低的远缘物种之间的序列比对[6]。
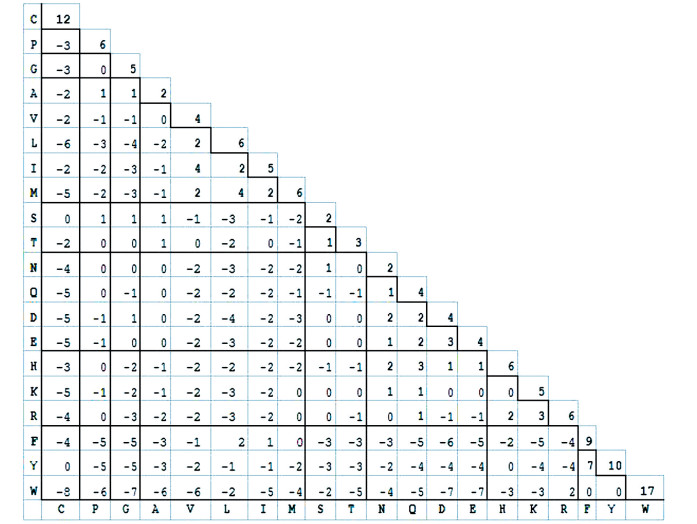
3.3 PAM250除BLOSUM系列计分矩阵外,PAM系列计分矩阵也是蛋白质序列比对时常用的计分矩阵。下面,我们以PAM250为例,说明其特点。此矩阵原始数据从以下网站下载(http://rosalind.info/glossary/pam250),本文做了改编,改编原则与BLOSUM62相同。
与BLOSUM62类似,PAM250的匹配分值均为正值,而错配分值绝大部分为零或负值。仔细分析这两个计分矩阵可以发现,无论是对角线上的匹配分值,还是同组或不同组氨基酸之间的错配分值,这两个矩阵之间都很不相同。BLOSUM62的最大匹配分值为11(色氨酸W),同为色氨酸W,PAM250的匹配分值为17。BLOSUM62错配分值范围为3到-4,而PAM250错配分值的范围为7到-8,远比BLOSUM62大。总之,无论是匹配分值还是错配分值,PAM250比BLOSUM62范围大。实际使用时,PAM250适用于相似性较低的序列之间的比对,具有较高灵敏度[7]。
PAM系列计分矩阵的英文全称为Point Accepted Mutation,即位点可接受突变矩阵,于上世纪七十年代构建,包括PAM10、PAM20、PAM30,一直到PAM500,共五十个矩阵(ftp://ftp.ncbi.nlm.nih.gov/blast/matrices)。与BLOSUM计分矩阵类似,PAM计分矩阵的适用范围也有差别,PAM10、PAM20等数字较小的矩阵,适用于相似性较高的序列之间的比对,而PAM250及其以上的矩阵,适用于相似性较低的序列之间的比对。需要注意的是,由于构建方法不同,这两个矩阵系列的数字互不对应。根据经验,PAM70与BLOSUM62对应,PAM30与BLOSUM90对应,而PAM250则于BLOSUM30对应。也就是说,实际比对时,分别采用两种对应的矩阵,比对结果比较接近(见表 5)。
| 表 5 相似性计分矩阵PAM250 Table 5 The PAM250 Scoring Matrix |
下面,以血红蛋白等为例,介绍双序列比对的具体应用,包括比对平台和工具选择、参数设置和结果分析等。
血红蛋白(Hemoglobin, HB)是重要生物大分子,在生命科学研究历史中具有特殊地位。上世纪五十年代,英国剑桥医学分子生物学实验室佩鲁茨(Max Perutz)研究组测定了抹香鲸血红蛋白的三维空间原子坐标结构,为生物大分子结构功能关系研究奠定了基础。人类基因组中有alpha和beta两大类血红蛋白编码基因,共编码9种不同血红蛋白。成人血液中的血红蛋白是异源四聚体,由两个alpha亚基和两个beta亚基组成(见图 3)。alpha亚基全长142 AA,beta亚基长度为147 AA。两个亚基结构上具有一定相似性,每个亚基结合一个血色素[9]。

|
图 3 人源血红蛋白分子三维空间结构 Figure 3 Three-dimensional structure of human hemoglobin |
斑头雁(Bar-headed Goose)和灰雁(Greylag Goose)同属于雁形目、鸭科、雁属。斑头雁为典型的候鸟,冬天在印度平原栖息而夏天在青海湖繁育后代,春秋两季则由南往北或由北往南迁徙,飞越喜马拉雅山; 而同为雁属(Anser)的灰雁则常年生活在印度平原。佩鲁茨在《分子生物学和演化》(Molecular Biology and Evolution)杂志创刊号上发表的开卷篇综述中推断,这两种雁的生活习性差别如此之大,可能与它们的血红蛋白序列和结构有关[10]。为此,我们可以对它们的序列进行比对。此处,我们采用EMBOSS Explorer分析平台。具体操作步骤如下。
1) 打开EMBOSS Explorer分析平台:
https://www.bioinformatics.nl/emboss-explorer
在程序导航菜单中找到整体比对ALIGNMENT GLOBAL,点击程序名needle。
2) 在UniProt数据库检索框中输入斑头雁alpha血红蛋白序列条目名HBA_ANSIN,点击Search按钮,在输出结果页面中点击页面上方Format按钮,选择FASTA格式,将斑头雁血红蛋白序列拷贝粘贴到needle程序第一个输入框中。
3) 在UniProt数据库检索框中输入灰雁alpha血红蛋白序列条目名HBA_ANSAN,将灰雁alpha血红蛋白序列拷贝并粘贴到needle程序第二个输入框中。
4) 选择默认计分矩阵EBLOSUM62(EMBOSS软件包中所有计分矩阵都冠以字母E)、默认起始空位(GAP OPEN)罚分10和延伸空位(GAP EXTEND)罚分0.5,序列末端空位(END GAP PENALTY)不予罚分(No)。
5) 点击Submit按钮,几秒钟后,输出运行结果。

|
输出结果显示,斑头雁和灰雁血红蛋白alpha亚基共有三个位点差异,其中一个位点为第119位,该位点灰雁为脯氨酸,而斑头雁为丙氨酸,即发生了P119A的突变。上世纪九十年代,北京大学研究团队测定了这两种雁血红蛋白的晶体结构,发现斑头雁血红蛋白alpha亚基第119位脯氨酸突变为丙氨酸,提高了斑头雁结合氧气的能力,证实了佩鲁茨的推断。
4.3 人和小鼠血红蛋白序列比对以上斑头雁和灰雁alpha血红蛋白仅差3个位点,比对结果一目了然。而人和小鼠分别属于哺乳纲灵长目和啮齿目,两者alpha血红蛋白差异位点较多。下面,我们利用EBI部署的EMBOSS软件包中needle程序,以人和小鼠alpha血红蛋白为例进行比对,具体操作步骤如下。
1) 打开EBI部署needle程序用户界面, 选择Protein序列比对:
https://www.ebi.ac.uk/Tools/psa/emboss_needle
2) 在UniProt数据库检索框中输入人alpha血红蛋白序列条目名HBA_HUMAN,点击Search按钮,在输出结果页面中点击页面上方Format按钮,选择FASTA格式,将人alpha血红蛋白序列拷贝粘贴到needle程序第一个输入框中。
3) 在UniProt数据库检索框中输入小鼠alpha血红蛋白序列条目名HBA_MOUSE,将小鼠alpha血红蛋白序列拷贝并粘贴到needle程序第二个输入框中。
4) 选择默认计分矩阵EBLOSUM62; 选择默认起始空位(GAP OPEN)罚分10和默认延伸空位(GAP EXTEND)罚分0.5,末端空位(END GAP PENALTY)不予罚分(False)。
5) 点击Submit按钮,几秒钟后,输出运行结果。
输出结果包括三部分,第一部分为所用程序名needle和运行日期,所选参数计分矩阵BLOSUM62和空位罚分10和0.5等。第二部分为相同位点(Identity)比例122/142和百分比85.9%、相似位点(Similarity)比例131/142和百分比92.3%、空位(Gaps)比例0/142和百分比0.0%,以及比对分值(Score)648.0。第三部分是具体比对结果。限于篇幅,下面仅列出比对结果。

|
比对结果显示,人和小鼠alpha血红蛋白绝大部分位点相同,用竖杠“|”表示; 不同位点则用句号“.”表示; 而用冒号“: ”表示的位点则称相似位点。所谓相似位点,是指性质相似的两种氨基酸,如苏氨酸T和丝氨酸S,缬氨酸V和异亮氨酸I,谷氨酸E和门冬氨酸D等。
4.4 人和小鼠血红蛋白编码区序列比对采用上述相同方法和步骤,将大鼠(HBA_RAT)和小鼠alpha血红蛋白进行序列比对。结果表明,两者之间的相同位点为120 AA,低于人和小鼠alpha血红蛋白之间相同位点数122 AA。查阅物种分歧时间网站(http://www.timetree.org)发现,人和小鼠分歧时间约9千万年,而大鼠和小鼠的分歧时间仅2千5百万年。我们知道,物种分歧年代越久远,积累的突变位点越多,序列相似性越低。上述人/小鼠、大鼠/小鼠alpha血红蛋白比对结果和预期相反,需要从分子生物学、遗传学和结构生物学等方面探其究竟,而密码子简并性是一个可能的原因。为证实这一推测,我们可以利用needle程序比较这三个物种alpha血红蛋白编码序列之间的相似性。具体步骤如下。
1) 打开EMBOSS Explorer分析平台,找到needle程序:
https://www.bioinformatics.nl/emboss-explorer
2) 打开NCBI核酸序列数据库网站:
https://www.ncbi.nlm.nih.gov/nucleotide
3) 输入登录号NM_000558.5,即可得到人的alpha血红蛋白mRNA序列,点击Send to按钮,选择Coding Sequences,即可下载编码区序列。
4) 按照上述方法下载小鼠alpha血红蛋白mRNA(登录号NM_008218.2)编码区序列。
5) 将上述人和小鼠alpha血红蛋白编码区序列分别粘贴到两个输入框中。
6) 点击参数选择按钮“More options”,将默认起始空位罚分10改为15,点击Submit按钮,即可得到运行结果(此处仅显示5’端150个碱基比对结果,其余省略)。
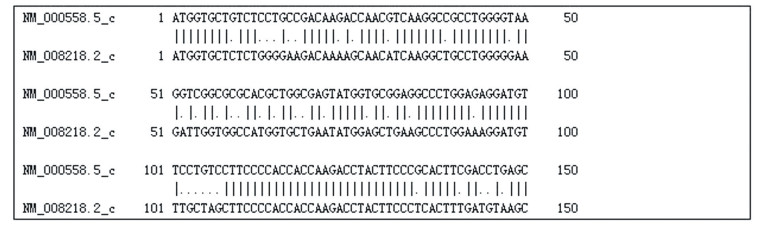
|
步骤6将默认起始空位罚分10改为15,可限制空位插入。若不做修改,则比对结果中有几处空位插入,其中有的空位插入到密码子中间,影响结果准确性。
按上述方法,比较大鼠alpha血红蛋白mRNA(登录号NM_013096)和小鼠alpha血红蛋白编码区序列。结果表明,人和小鼠alpha血红蛋白mRNA编码区序列共有350个相同位点(81.6%),而大鼠和小鼠alpha血红蛋白mRNA序列编码区共有383个相同位点(89.3%)。显然,同为啮齿目的小鼠和大鼠之间alpha血红蛋白编码区序列相似性较高。
以上人和小鼠、大鼠和小鼠alpha血红蛋白序列比对给我们一定启示。有些重要功能基因在近缘物种中的直系同源基因,其序列相似性较高,仅用蛋白质序列进行比对分析,结果不一定可靠,需要同时比较其编码基因的DNA或mRNA序列。
4.5 alpha和beta血红蛋白序列比对以上实例所比对的两个序列长度相同,相似性较高。下面以人的alpha和beta血红蛋白为例,介绍长度不同、差异较大的两个序列之间的比对。据报道,alpha和beta两个血红蛋白基因家族源自哺乳动物祖先全基因组复制,后经染色体局部区域复制,又各自产生了多个基因。经过4亿多年的演化,包括突变、插入和删除,两者之间积累了相当多的差异位点,包括某些位点的插入和缺失,但依然保留一定的序列相似性。下面,我们利用NCBI提供的BLAST分析平台中基于Needleman-Wunsch整体比对算法的Global Align,介绍人的alpha血红蛋白HBA_HUMAN和beta血红蛋白HBB_HUMAN的序列比对。
1) 打开NCBI数据库相似性搜索平台BLAST:
https://blast.ncbi.nlm.nih.gov/Blast.cgi
在页面下方专用搜索程序(Specialized searches)所列工具中选择基于Needleman-Wunsch算法的整体比对程序Global Align,选择蛋白质(Protein)序列比对。
2) 在两个输入框中分别输入人alpha血红蛋白(HBA_HUMAN)和beta血红蛋白(HBB_HUMAN)的UniProt数据库登录号P69905和P68871。
3) 点击BLAST进行比对,在输出结果页面中点击Alignments标签,显示序列比对细节,点击点阵图Dot Plot标签,以图形方式显示比对结果。
比对结果以表格方式输出总分值(NW Score)282,相同位点(Identities)比例65/149和百分比44%,正分值(Positives)比例90/149和百分比60%,以及空位(Gaps)比例9/149和百分比6%。以文本方式输出具体比对结果。

|
比对结果中,Query为alpha血红蛋白P69905,长度为142 AA,Sbjct (Subject缩写) 为beta血红蛋白,长度为147 AA。中间一行表示匹配情况,相同位点用该位点氨基酸表示,分值为正的位点用加号“+”表示。两个序列之间共有四处空位插入,alpha血红蛋白有三处,其中两处插入一个空位,另一处插入5个空位; beta血红蛋白有一处长度为2的空位。
除文本方式输出比对结果外,还可以用可视化的点阵图方式查看结果,alpha血红蛋白(P69905)位于X轴,beta血红蛋白(P68871)位于Y轴(见图 4)。

|
图 4 人alpha和beta血红蛋白双序列BLST比对点阵图 Figure 4 Dot plot of BLAST output between human alpha and beta hemoglobin |
主对角线上三处向上跳跃的缺口即为alpha血红蛋白的三个空位插入区域; 向右跳跃的缺口则是beta血红蛋白的一个空位插入区域。显然,和上面的几个实例相比,人的alpha和beta血红蛋白差异较大,相似性位点约占一半左右。尽管如此,比对结果中具有多处保守区域或相同位点,这就决定了两者三维空间结构也十分相似。
5 实例2:抗菌肽和多肽毒素 5.1 研究背景以上我们以血红蛋白为例,对几种不同的双序列比对方法做了简单介绍。这些实例中,所比对的序列相似性较高,两者之间具有明显的保守区域或保守位点。下面我们以抗菌肽和多肽毒素为例,说明相似性较低的序列之间的序列比对所采用的策略,特别是改变计分矩阵和空位罚分对比对结果的影响。
2000年,北京大学生命科学学院吴光耀、赵进东课题组从产于我国云南的草药美洲商陆(Pokeweed, Phytolacca americana)种子中分离到一种具有抑制真菌和革兰氏阳性菌等微生物生长活性的多肽,称抗菌肽(Antimicrobial Peptide, AMP)。AMP序列全长38 AA,含6个半胱氨酸[11]。AMP的三维空间结构为典型的“三桥三叠”折叠模式,即三对二硫键和三个beta折叠。
这种三桥三叠的折叠模式在蜘蛛毒素、芋螺毒素等小分子量多肽毒素中较为常见,二硫键的配对方式多为1-4, 2-5, 3-6,即第1个和第4个半胱氨酸配对,第2个和第5个半胱氨酸配对,第3个和第6个半胱氨酸配对。九十年代初,湖南师范大学梁宋平教授从我国广西地区特有的虎纹捕鸟蛛毒液中分离纯化得到的虎纹捕鸟蛛毒素-I(Huwentoxin-I, HWT1)。HWT1序列全长33 AA,也含6个半胱氨酸,三维空间结构和二硫键配对方式与抗菌肽AMP完全相同。
除三对保守的二硫键外,AMP和HWT1的序列相似性很低。对于这类富含半胱氨酸的小分子量多肽,采用默认计分矩阵和空位罚分难以得到理想的结果,需要根据实际情况适当调整参数。下面,我们以AMP和HWT1为例,说明不同计分矩阵和空位罚分对比对结果的影响。
5.2 默认计分矩阵和默认空位罚分抗菌肽AMP和虎纹捕鸟蛛毒素-I HWT I的三维空间结构都已通过核磁共振方法测定,PDB数据库中的登录号分别为1DKC和1QK6,其序列数据可从PDB数据库中下载。此处,我们采用EMBOSS Explorer分析平台。具体操作步骤如下。
1) 打开EMBOSS Explorer分析平台, 在左侧程序导航菜单中找到整体比对ALIGNMENT GLOBAL栏目,点击该栏目下程序名needle:
https://www.bioinformatics.nl/emboss-explorer
2) 打开蛋白质结构数据库PDB:
3) 在检索框中输入抗菌肽登录号1DKC,在Display Files下拉式菜单中选择FASTA Sequence,复制FASTA格式序列,粘贴到程序EMBOSS Explorer分析平台needle程序第1个输入框中,删除序列注释信息,只保留登录号1DKC。
4) 按上述同样方法,将虎纹捕鸟蛛毒素-I(登录号1QK6)序列复制、粘贴到第2个输入框中,删除序列注释信息,只保留登录号1QK6。
5) 采用默认计分矩阵EBLOSUM62,默认起始空位罚分10.0、延伸空位罚分0.5。
6) 点击Run needle,即可得到比对结果。
与EBI部署的needle程序相同,输出结果中给出运行日期,计分矩阵和空位罚分等基本信息,以及相同位点、相似位点、空位所占比例和百分比及比对分值,下面仅列出比对结果。

|
分析上述比对结果可以发现,蜘蛛毒素1QK6的N-端有多个空位插入,其第1个半胱氨酸与抗菌肽1DKC的第2个半胱氨酸匹配,而第2个和第5个半胱氨酸无法与蜘蛛毒素1QK6的第2和第5个半胱氨酸匹配。显然,这个比对结果与实际情况相去甚远。
5.3 调整计分矩阵为此,我们尝试改变计分矩阵和空位罚分,以图改善比对结果。本文第3节“常用计分矩阵”介绍了BLOSUM和PAM两个系列的计分矩阵,BLOSUM62为EMBOSS软件包的默认计分矩阵,适用于大部分情况。PAM系列计分矩阵中,PAM250适用于相似性较低的序列比对,此处不妨可以试一下。具体操作步骤如下。
1) 在EMBOSS Explorer分析平台中,回退到上述输入界面。
2) 在计分矩阵选择框中,输入计分矩阵PAM250的文件名EPAM250。
3) 点击Run needle,即可得到如下比对结果。

|
上述结果解决了第1和第2个半胱氨酸匹配问题,但蜘蛛毒素1QK6的第5个半胱氨酸仍没有准确匹配,两个序列的C-末端各有一处连续空位。适当降低空位罚分值,有可能改变输出结果。
5.4 调整计分矩阵和空位罚分为此,我们在改变计分矩阵的基础上,适当降低空位罚分。具体操作步骤如下。
1) 在EMBOSS Explorer分析平台中,回退到上述输入界面。
2) 在计分矩阵选择框中,输入计分矩阵EPAM250。
3) 在空位罚分选择框中,输入起始空位罚分值5。
4) 点击Run needle,即可得到如下比对结果。
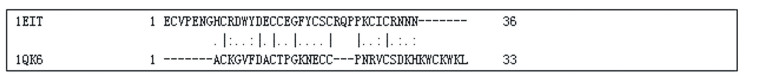
|
比对结果中6个半胱氨酸都得到了正确匹配。与前两次相比,这一结果跟接近实际。
5.5 调整末端空位罚分有时,双序列比对结果中会出现末端连续几个空位插入的情况,影响整体比对结果。从PDB网站下载芋螺毒素1EIT序列,删除C-末端未定氨基酸X,采用BLOSUM62默认计分矩阵、默认起始空位罚分为10、延伸空位罚分为0.5时,芋螺毒素1EIT和蜘蛛毒素比对结果如下。

|
比对结果中蜘蛛毒素N-端和芋螺毒素的C-端都有连续空位,结果不太理想。EMBOSS软件包中的needle程序可由用户设定末端空位罚分。具体操作步骤非常简单,只要将EMBOSS Explorer末端空位(Apply end gap penalty)下拉菜单中的No改为Yes即可。采用上述默认计分矩阵和默认空位罚分,设定末端空位罚分后的比对结果如下。

|
显然,设置末端空位罚分后,比对结果更加合理。
6 实例3:植物特异转录因子SBP 6.1 研究背景我们知道,转录调控是真核生物基因表达调控的重要环节。转录因子(Transcription Factor, TF)与其靶标基因启动子区域顺式元件特异结合,控制基因在不同组织、不同发育阶段、不同环境条件下表达,是转录调控的分子基础。根据DNA结合结构域的序列特征,转录因子分为不同家族[12]。
Squamosa Promoter-binding Protein (SBP)是一个植物特异转录因子家族。1996年,德国植物育种研究所Huijser实验室从金鱼草(Antirrhinum majus)中克隆到两个基因SBP1和SBP2[13]。之后不久,又在拟南芥、玉米等植物中克隆到多个类SBP家族成员,因其中大部分尚未进行实验鉴定,故称类SBP转录因子(SBP-like, SPL)。
2008年,我们利用数据库检索和序列相似性搜索等方法,搜集了拟南芥、水稻、裸子植物、蕨类、苔藓和绿藻等植物代表性谱系120个SBP转录因子成员,进行了基因结构、保守结构域等分析,构建了系统发生树,推测了SBP基因家族的起源和演化。近年来,随着基因组测序不断普及,许多植物的基因组已经测定,西北农林大学、南京农业大学、北京林业大学等鉴定了棉花、花生、辣椒、苹果、葡萄、毛竹、丹参、菊花等重要经济作物、水果、花卉中的SBP转录因子。2019年更新的植物转录因子数据库(http://planttfdb.gao-lab.org)中收录了165个物种共4168个SBP家族转录因子。
SBP转录因子家族具有许多重要生物学功能,包括花和果实发育、孢子发育、激素应答、抗霉菌侵蚀等。玉米SBP转录因子LG1缺失突变体不能形成叶舌和叶耳。野生玉米果实有颖壳包裹,而栽培玉米果实无外壳包裹。这种表型变化主要由玉米果实形态相关基因控制。该基因为SBP转录因子家族成员,野生玉米第6位氨基酸为赖氨酸Lys,而栽培玉米中为门冬酰氨Asn。西红柿果实成熟关键基因SBP启动子区域甲基化修饰突变体可抑制果实成熟。水稻OsSPL7,OsSPL13, OsSPL14, OsSPL16和OsSPL17调控植株分蘖数、谷粒数量和大小。
UniProt蛋白质数据库中收录了金鱼草SBP1和SBP2的序列及其注释信息,序列条目名分别为SBP1_ANTMA和SBP2_ANTMA,下划线前为基因名,下划线后为金鱼草物种名缩写,由属名Antirrhinum 前三个字母ANT和种名majus前两个字母MA组成。这两个序列长度分别为131 AA和171 AA,相差40个氨基酸。
下面,我们以金鱼草SBP1和SBP2为例,说明整体比对和局部比对两种不同方法,以及计分矩阵和空位罚分等参数对比对结果的影响,说明不同方法的适用范围。并比较采用动态规划的Smith-Waterman局部比对和采用启发式算法的BLAST局部比对所得结果的差别。
6.2 整体比对从UniProt蛋白质数据库主页面检索框中分别输入序列条目名SBP1_ANTMA和SBP2_ANTMA,用FASTA格式提取序列,用EBI部署的EMBOSS软件包中整体比对程序needle进行比对,采用默认计分矩阵EBLOSUM62、默认起始空位罚分10和默认延伸空位罚分0.5(具体步骤可参阅4.4人和小鼠alpha血红蛋白比对),比对结果如下。
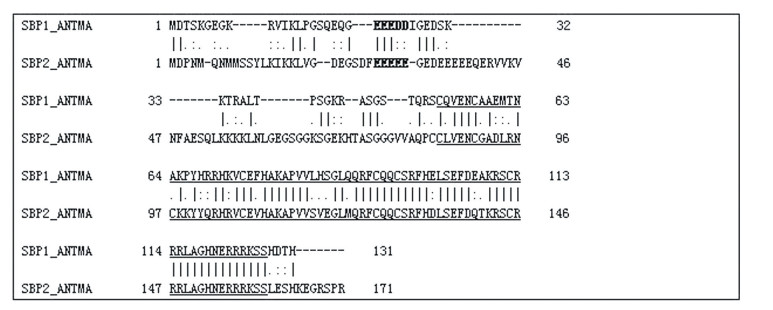
|
分析比对结果可以发现,这两个序列长度相差40 AA,SBP1的N-端出现大片段连续空位,与SBP2的序列相似性较低; 从52位C开始一直到127位S共计76个氨基酸(下划线标记),两者相似性很高。实际上,相似性较高的保守区域就是SBP转录因子家族特有的DNA结合区域,与下游靶基因Squamosa(SQUA)启动子区域顺式元件特异结合。该靶基因SQUA编码另外一个转录因子,属于MADS家族,调控花发育。据文献报道,SBP转录因子的核定位信号为两个连续的碱性氨基酸片段RRR和RRRK(粗体标记),位于DNA结合结构域的C-末端。此处需要说明,上述结果中下划线和粗体并非程序输出结果,而是分析后添加的。
进一步分析上述输出结果发现,SBP1 N-端有EEEDD连续五个带负电氨基酸,而SBP2则有EEEEE和EDEEEEE两个连续带负电氨基酸片段(粗体标记),中间只隔了一个氨基酸G。采用较低的起始空位罚分5,其它参数不变,则输出结果有所不同,可以看出这两个序列中带负电的片段EEEDD和EEEEE具有较好的匹配。

|
整体比对适用于两个序列长度相差不大、相似性较高的同源蛋白之间的比对。例如,拟南芥SPL3_ARATH长度与金鱼草SBP1相同,也是131 AA。整体比对结果表明,这两个序列的相同位点占序列全长58.6%,而相似位点高达70.7%。进一步分析可以发现,两者C-端相似性极高,说明其DNA结合结构域序列相当保守。

|
上述needle比对结果,从整体上提供了金鱼草SBP1和SBP2蛋白质序列之间的保守区域和差异区域。下面,我们采用基于Smith-Waterman动态规划算法的局部比对,分析比对结果是否有所不同。
EBI部署的EMBOSS软件包中,包括局部比对程序water。打开该程序用户界面,从UniProt中分别提取SBP1和SBP2的FASTA格式序列,粘贴到输入框中,选择默认计分矩阵EBLOSUM62和空位罚分10/0.5,结果如下。
从比对结果可以看出,局部比对与整体比对的主要差别在于局部比对只考虑两个序列中相似性较高的区域。若序列两端相似性较低,不在比对结果中列出,即比对结果中不包括SBP2第1-33位和第165-171位序列。局部比对多用于长度差异较大的两个序列,如拟南芥SBP转录因子家族共有17个成员,其序列长度差很大,最短的SPL3仅131个氨基酸,而最长的SPL14由1035个氨基酸组成,用局部比对更容易找出两者之间的保守区域DNA结合结构域。

|
上述基于Smith-Waterman动态规划算法的water局部比对结果,能较好地找出SBP1和SBP2之间的相似区域,而采用基于启发式算法的BLAST程序,也可以找出两者之间的相似区域,结果稍有不同。下面,我们仍以金鱼草SBP1和SBP2为例,说明比对步骤和输出结果。
1) 打开NCBI数据库相似性搜索平台BLAST:
https://blast.ncbi.nlm.nih.gov/Blast.cgi
2) 选择蛋白质序列比对Protein BLAST,即blastp。
3) 勾选双序列比对选择框(Align two or more sequences)。
4) 在两个输入框中分别输入金鱼草SBP1_ANTMA和SBP2_ANTMA序列。
5) 点击BLAST进行比对。
输出结果页面给出所用程序和所选参数,并以图表方式显示比对结果概要,以文本方式给出比对结果细节。此处,我们选择默认参数,即字长为3,期望值为0.05,计分矩阵为BLOSUM62,起始空位罚分为11,延伸空位罚分为1。比对结果如下。
与water比对结果比较,BLAST比对结果更加突出DNA结合结构域高度保守区域
7 实例4:癌胚抗原 7.1 研究背景癌胚抗原(Carcinoembryonic Antigen, CEA)是一种细胞表面糖蛋白,多在直肠癌、胃癌等恶性肿瘤中表达,临床上常作为非特异性肿瘤标记物和肿瘤化疗预后指标。人的癌胚抗原基因家族包括两个亚家族,其中CEA亚家族共有12个不同成员(见图 5),所编码的蛋白质属免疫球蛋白超家族成员(http://www.carcinoembryonic-antigen.de/)。所有成员N-端均含长度为34 AA的信号肽(Signal Peptide),第35位开始则为免疫球蛋白可变结构域(Immunoglobin Variable Domain, IgV),长度约为110个氨基酸。除可变结构域外,有的成员还含一个或多个免疫球蛋白恒定结构域(Immunoglobin Constant Domain, IgC)。不同成员之间可变结构域序列相似性较高; 恒定结构域分A和B两种亚型,不同成员之间相同亚型恒定结构域也有较高相似性[14]。

|
图 5 人癌胚抗原CEA5和CEA6结构域 Figure 5 Domain structure of human carcinoembryonic antigen CEA5 and CEA6 |
下面,我们以CEA家族中两个成员CEA5和CEA6为例,说明如何利用不同比对方法,寻找保守性结构域。UniProt蛋白质数据库中收录了CEA5和CEA6的蛋白质序列,并在家族和结构域(Family & Domains)注释信息中给出结构域名称和位置。人的CEA5序列条目名为CEAM5_HUMAN,序列全长702AA,N-端1-34为信号肽,C-端676-702为膜结合序列模体。第35-144为可变型免疫球蛋白结构域(Ig-like V-type),第145-675含6个免疫球蛋白恒定结构域(Ig-like C2-type)。根据序列相似性,这6个恒定结构域分A和B两种类型。人的CEA6序列条目名为CEAM6_HUMAN,全长344 AA,N-端1-34为信号肽,C-端315-344为膜结合序列模体,第35-144为可变型免疫球蛋白结构域,第145-314含2个免疫球蛋白恒定结构域,分A型和B型两种类型。
7.2 BLAST查找多个结构域利用needle进行整体序列比对可以发现,这两个序列长度差别很大,N-端序列相似性较高。而用water进行局部序列比对,可以分别找出CEA6中可变结构域N和两个恒定结构域A和B与CEA5中对应的结构域。比对结果表明,CEA6中恒定结构域A(145-232)与CEA5中的恒定结构域A3 (501-588)相似性最高; 而CEA6恒定结构域B(145-232)与CEA5的恒定结构域B3 (501-588)相似性最高。也就是说,基于动态规划算法的局部比对程序water的比对结果,每次只能找出相似性最高的区域。如果需要同时找出CEA6中的恒定结构域A与CEA5中哪几个恒定结构域具有相似性,则可以采用BLAST双序列比对。
1) 打开NCBI数据库相似性搜索平台BLAST:
https://blast.ncbi.nlm.nih.gov/Blast.cgi
2) 选择蛋白质序列比对Protein BLAST,即blastp。
3) 勾选双序列比对选择框(Align two or more sequences)。
4) 在第一个输入框中输入CEA6的UniProt数据库登录号P40199,并输入比对范围From 145 To 232。
5) 在第二个输入框中输入CEA5的UniProt数据库登录号P06731,并输入比对范围From 145 To 675。
6) 打开参数选择(Algorithm Parameters)窗口,将默认字长(Word Size)3改为6。
7) 点击BLAST进行比对。
查看比对结果可以发现,CEA6中恒定结构域A在CEA5中共有三个相似性结构域,第一个与water比对结果一样,为CEA5中的A3结构域,相同位点比例为75/88 (85%); 第二个为CEA5中A1结构域,相同位点比例为71/88 (81%); 第三个为CEA5中A2结构域,相同位点百分比为70/88 (80%)。上述比对步骤6将字长改为6,可以提高比对结果的特异性(Specificity),降低假阳性(False Positive)。
7.3 Dotlet寻找重复结构域以上比对结果表明,CEA5的三个恒定结构域A1, A2和A3与CEA6的恒定结构域A都具有较高相似性。按此推测,CEA5的这三个恒定结构域之间也有较高的序列相似性。换句话说,CEA5中存在重复结构域,即重复序列(Repeat Sequence)片段或重复区域。实际上,从UniProt数据库的注释信息中也可以发现,CEA5的这三个结构域具有较高序列相似性,而另外三个结构域B1, B2和B3之间也具有较高相似性。
利用瑞士生物信息研究所开发的点阵图分析平台Dotlet,即可找出序列内部的重复区域。具体步骤如下。
1) 打开Dotlet分析平台网站:
2) 点击页面上方SEQUENCE1标签,将UniProt数据库中提取的CEAM5_HUMAN序列粘贴到输入框中,删除第一行注释信息,只保留序列本身。
3) 点击页面上方SEQUENCE2标签,将上述SEQUENCE1输入框中的序列粘贴到SEQUENCE2输入框中,删除第一行注释信息,只保留序列本身。
4) 左右移动页面中部直方图下两个滑动球,以获得最佳信噪比。
5) 左右移动页面下方序列窗口上下两个滑动球,将十字型红色指示标线交叉点置于主对角线下第一条平行线起始处。
分析结果以图形方式显示(见图 6)。可以看到主对角线左下方和右上方各有两条对称的平行线。参阅CEA5结构域分布方式(见图 5)可以发现,较长的一对平行线表明从A2到B3四个结构域与从A1到B2的四个结构域序列相似,而较短的一对平行线表明从A3到B3两个结构域与从A1到B1的两个结构域序列相似。

|
图 6 点阵图程序Dotlet显示癌胚抗原CEA5中的重复序列 Figure 6 Repeat sequence in carcinoembryonic antigen CEA5 revealed by Dotlet platform |
我们知道,序列比对的前提是基于分子生物学基本规则“序列决定结构、结构决定功能”; 换而言之,具有共同祖先的两个序列,无论是直系同源或者是旁系同源,经过亿万年的演化和物种分化,尽管某些位点发生了替换、插入或删除,两者仍保持一定相似性,其结构和功能也基本相同或相似。然而,由趋同进化机制产生的两个基因,其编码的蛋白质结构很可能十分相似,并具有类似的功能,但其序列却大相径庭。
例如,分布于流感病毒被膜的神经氨酸酶(Neuraminidase)是一种糖蛋白,能够水解唾液酸与宿主细胞血凝素之间的糖苷键,因此又称唾液酸酶(Cytosolic Sialidase)。显然,神经氨酸酶在成熟病毒颗粒逃离宿主细胞而感染新细胞的过程中具有关键作用。2003年,H5N1甲型禽流感爆发,并很快感染人,成为当时主要流行性感冒。瑞士罗氏公司生产的流感病毒神经氨酸酶抑制剂达菲(Oseltamivir)上市,用于治疗甲型流感和季节性流感。之后不久,日本等部分亚洲国家报道了上千达菲副作用案例,主要症状为精神恍惚和皮肤过敏,甚至出现个别服用达菲的中学生跳楼自杀的极端案例。
2007年北京大学生物信息中心魏丽萍教授课题组利用生物信息学方法,结合酶活测定实验结果,分析了流感药物达菲产生副作用的可能机制[15]。研究表明,人的唾液酸酶和流感病毒唾液酸酶的三维空间结构非常相似,尤其是和底物结合的活性中心,具有几乎相同的空间结构,结合底物的方式也基本相同。通过搜索NCBI的单核苷酸多态性(Single Nucleotide Polymorphism, SNP)数据库dbSNP,发现日本等亚洲人群的唾液酸酶第41位产生了R41Q非同义突变,使得达菲更加容易进入人的唾液酸酶的活性中心,从而抑制了该酶的正常作用。
8.2 结构和序列比对对于这种两者结构均已经测定的蛋白质,则可以用蛋白质结构数据库PDB网站提供的结构比对工具。该工具不仅可用于比较两个蛋白质之间三维空间结构的异同,同时也给出这两个蛋白质序列之间的差异。可以通过PDB数据库网站提供的结构比对工具,比较流感病毒和人的唾液酸酶结构,同时给出序列比对信息。具体操作步骤如下:
1) 打开蛋白质结构数据库PDB网站:
2) 在分析工具(Analyze)下拉菜单中选择成对结构比对(Pairwise Structure Alignment),点击页面中蛋白质结构比对工具(Protein Structure Alignment tool)或输入以下网址启动比对页面:
https://www.rcsb.org/alignment
3) 在第一个PDB ID输入框中输入流感病毒唾液酸酶登录号2BAT,在Chain ID中选择A链。
4) 在第二个PDB ID输入框中输入人的唾液酸酶登录号1VCU,在Chain ID中选择A链。
5) 选择默认比对方法jFATCAT (Rigid)和默认比对参数,点击Compare按钮进行比对。
分析比对结果可以发现,流感病毒和人的唾液酸酶结构非常相似,而序列却没有任何相似性。对于这样的实例,常规的比对方法无能为力,无论是整体比对或者是局部比对,无论是动态规划还是启发式算法,都无法得到准确的比对结果。
9 结语本文所举实例选自笔者参与或合作的研究课题,并用于为北京大学生命科学学院和中国农业科学院研究生院开设的“实用生物信息技术”研究生课程(Applied Bioinformatics Course, ABC, http://abc.gao-lab.org)教学[16]。选修本课程的同学,多为从事分子生物学和遗传学等“湿实验”的低年级硕士或博士研究生。细心的读者可以发现,本文主要介绍双序列比对如何用于正在进行的研究课题中遇到的实际问题,而不是双序列比对的原理和方法,即重在实际操作。
双序列比对的分析平台和软件工具很多,本文主要介绍NCBI和EBI等国际著名生物信息中心基于浏览器的程序。根据笔者多年来教学实践中的经验,这些分析平台用户界面友好、操作方便、结果显示清晰。需要说明的是,这些分析平台的用户界面和输出格式会不定期更新。
本文旨在通过这些实例,使读者特别是分子生物学研究领域从事“湿实验”的年轻读者对双序列比对方法和平台选择有所了解。例如,什么情况下采用整体比对、什么情况下采用局部比对、什么情况下采用点阵图; 并能分清动态规划和启发式算法的区别,遇到具体问题时选择适当的程序。建议读者先把本文介绍的实例作为练习,按本文介绍的步骤进行实际操作,并在掌握操作方法和理解操作过程的基础上,适当改变计分矩阵和空位罚分等程序参数,分析不同参数对输出结果的影响。在顺利完成文中所举实例后,结合研究课题相关或自己熟悉的例子,尝试参阅不同的例子,进行实际操作,有时需要采用整体比对和局部比对等几种不同方法,才能得到满意的结果。
本文介绍的分析平台均在国外,并且分布在几个不同的生物信息中心。由于网络带宽等因素限制,一定程度上影响了国内用户正常使用这些分析工具。值得庆幸的是,中国科学院北京基因组研究所(国家生物信息中心)正在开发的生物信息工具箱(https://ngdc.cncb.ac.cn/bit)计划将needle, water, BLAST等常用双序列比对程序部署到中心服务器上,为国内用户快速、高效使用这些工具提供方便。
本文仅讨论双序列比对,而多个序列同时进行比对也是分子生物学研究工作者经常遇到的问题。此外,本文所用的BLAST双序列比对只是BLAST数据库相似性搜索的一个特殊应用,而BLAST分析平台还有许多功能和具体应用。上述多序列比对和BLAST数据库搜索实例,计划另行介绍。
致谢 感谢北京大学生命科学学院和中国农业科学院研究生院多年来对“实用生物信息技术”研究生课程教学的支持。感谢北京基因组研究所鲍一明和章张研究员、先声医学诊断公司颜林林博士、新加坡国立大学周群飞博士,以及北京大学张晨妍、张文心、饶希晨、张雪昂、刘曦瑞、杨德昌、金录嘉和中国农业科学院邢宏阳、方荣民等同学的修改意见。
| [1] |
朱玉贤, 李毅, 郑晓峰, 等. 现代分子生物学[M]. 5版. 北京: 高等教育出版社, 2021. ZHU Yuxian, LI Yi, ZHENG Xiaofeng, et al. Modern molecular biology[M]. 5th ed. Beijing: Higher Education Press, 2021. (  0) 0) |
| [2] |
GASTEIGER E, HOOGLAND C, GATTIKER A, et al. Protein identification and analysis tools on the ExPASy Server[M]//WALKER J N. The Proteomics Protocols Handbook. [S.L.]: Humana Press, 2005, 571-607.
( 0) |
| [3] |
XU Zhongneng, YANG Yayun, HUANG Beibei. A teaching approach from the exhaustive search method to the Needleman-Wunsch algorithm[J]. Biochemistry and Molecular Biology Education, 2017, 45(3): 194-204. DOI:10.1002/bmb.21027 ( 0) |
| [4] |
SMITH T F, WATERMAN M S, FITCH W M. Comparative biosequence metrics[J]. Journal of Molecular Evolution, 1981, 18(1): 38-46. DOI:10.1007/BF01733210 ( 0) |
| [5] |
HENIKOFF S, HENIKOFF J G. Amino acid substitution matrices from protein blocks[J]. Proceedings of the National Academy of Sciences USA, 1992, 89(22): 10915-10919. DOI:10.1073/pnas.89.22.10915 ( 0) |
| [6] |
MOUNT D W. Using BLOSUM in sequence alignments[J]. CSH Protocols, 2008, 2008: 39. DOI:10.1101/pdb.top39 ( 0) |
| [7] |
MOUNT D W. Comparison of the PAM and BLOSUM amino acid substitution matrices[J]. CSH Protocols, 2008, 2008: 59. DOI:10.1101/pdb.ip59 ( 0) |
| [8] |
罗静初. EMBOSS软件包序列分析程序应用实例[J]. 生物信息学, 2021, 19(1): 1-25. LUO Jingchu. Application examples of EMBOSS sequence analysis program[J]. Chinese Journal of Bioinformatics, 2021, 19(1): 1-25. DOI:10.12113/202008002 ( 0) |
| [9] |
HARDISON R C. Evolution of hemoglobin and its genes[J]. Cold Spring Harbor Perspectives in Medicine, 2012, 2(12): a011627. DOI:10.1101/cshperspect.a011627 ( 0) |
| [10] |
JESSEN T H, WEBER R E, FERMI G, et al. Adaptation of bird hemoglobins to high altitudes: Demonstration of molecular mechanism by protein engineering[J]. Proceedings of the National Academy of Sciences USA, 1991, 88(15): 6519-22. DOI:10.1073/pnas.88.15.6519 ( 0) |
| [11] |
LIU Yingfang, LUO Jingchu, XU Chunyu, et al. Purification, characterization, and molecular cloning of the gene of a seed-specific antimicrobial protein from pokeweed[J]. Plant Physiology, 2000, 122(4): 1015-1024. DOI:10.1104/pp.122.4.1015 ( 0) |
| [12] |
靳进朴, 郭安源, 何坤, 等. 植物转录因子分类、预测和数据库构建[J]. 生物技术通报, 2015, 31(33): 68-77. JIN Jinpu, GUO Anyuan, HE Kun, et al. Classification, prediction and database construction of plant transcription factors[J]. Biotechnology Bulletin, 2015, 31(33): 68-77. DOI:10.13560/j.cnki.biotech.bull.1985.2015.07.001 ( 0) |
| [13] |
KLEIN J, SAEDLER H, HUIJSER P. A new family of DNA binding proteins includes putative transcriptional regulators of the Antirrhinum majus floral meristem identity gene SQUAMOSA[J]. Molecular and General Genetics, 1996, 250(1): 7-16. DOI:10.1007/BF02191820 ( 0) |
| [14] |
HAMMARSTROM S. The carcinoembryonic antigen (CEA) family: Structures, suggested functions and expression in normal and malignant tissues[J]. Seminars in Cancer Biology, 1999, 9(2): 67-81. DOI:10.1006/scbi.1998.0119 ( 0) |
| [15] |
LI Chuanyun, YU Quan, YE Zhiqiang, et al. A nonsynonymous SNP in human cytosolic sialidase in a small Asian population results in reduced enzyme activity: potential link with severe adverse reactions to oseltamivir[J]. Cell Research, 2007, 17(4): 357-362. DOI:10.1038/cr.2007.27 ( 0) |
| [16] |
罗静初. 实用生物信息技术课程教学实例[J]. 生物技术通报, 2015, 31(11): 102-111. LUO Jingchu. Teaching examples of applied bioinformatics course[J]. Biotechnology Bulletin, 2015, 31(11): 102-111. DOI:10.13560/j.cnki.biotech.bull.1985.2015.11.004.6 ( 0) |